Application of resonance to H2 Chemistry
This is where things get meaty. We see resonance in so many chapters – arenes, halogen derivatives, carboxylic acids, nitrogen compounds, and more. Resonance can be used to explain so many concepts! We can draw a quick map of four very commonly tested resonance concepts. Behold the four horsemen of resonance in H2 Chemistry:

Of course, these are not exhaustive. You may encounter other concepts involving resonance, but these are the four that are usually tested.
Disclaimer: concepts 1 and 2 are found in the chapter of halogen derivatives, concept 3 in the chapter of alcohols and carboxylic acids, and concept 4 in the chapter of nitrogen compounds. If you have not studied any of these chapters, I suggest you do so before you proceed! Do also read up on hybridisation first.
1. Partial double bond character
In the chapter of halogen derivatives, we encountered alkyl halides that undergo nucleophilic substitution. The nucleophilic substitution of an alkyl halide occurs when its halide is substituted by an incoming nucleophile. In the following example, the chloride is being substituted by a hydroxide nucleophile.

Nucleophilic substitution, however, does not take place readily in an aryl halide (an aryl halide is a molecule with a halogen atom directly bonded to a benzene ring).

We can explain the difference by examining the nucleophilic substitution process – nucleophilic substitution involves breaking the C–Cl bond in both the alkyl and aryl chloride. If the C–Cl bond could not be broken in the aryl chloride, it would mean that the C–Cl bond in the aryl chloride is stronger than the C–Cl bond in the alkyl chloride, thus requiring a lot more energy to break.
What makes the C–Cl bond stronger in an aryl halide? To which you might answer:

Yes, you’re right! But let’s elaborate on this answer. In benzene, every carbon is sp² hybridised. The Cl has at least one lone pair of electrons and is connected to a sp² hybridised carbon, and is therefore also sp² hybridised (see blog post on hybridisation). Therefore, we can draw the following p orbitals on every carbon and Cl in chlorobenzene:

Recall the two criteria that must be fulfilled for delocalisation to occur. (see blog post Resonance Explained)
Since
1. All the p orbitals are adjacent to each other and
2. All the p orbitals are aligned to each other,
The p orbital on Cl can overlap with the six p orbitals of the benzene ring (or the pi electron cloud of benzene), allowing the lone pair of electrons in the p orbital of Cl to delocalise into the pi electron cloud of benzene. When the lone pair of electrons on Cl delocalises into the p orbital of C1, the two electrons form a new bond between C1 and Cl – a partial double bond (indicated by the dotted line (------) in the diagram below). The partial double bond character of C–Cl strengthens the bond, making it difficult to break.
And that is why aryl halides do not undergo nucleophilic substitution reactions, as opposed to alkyl halides.

2. Stability of carbocations
Resonance can also be used to explain why a molecule preferably undergoes nucleophilic substitution through the Sₙ1 over the Sₙ2 mechanism.
Will the following molecule, 1-(chloromethyl)benzene, undergo Sₙ1 or Sₙ2?

Let us first examine the Sₙ1 and Sₙ2 mechanisms (without involving stereochemistry):
Sₙ2:

Sₙ1:

The main difference between the Sₙ1 and Sₙ2 mechanisms is that the Sₙ2 takes place in one step (note: the formation of the transition state is not counted as a step) while the Sₙ1 takes place in two steps.
Sₙ1 takes place when the carbocation formed is stable. The more stable the carbocation, the easier it is to form, the faster the rate of step 1 (rate-determining step), the faster the Sₙ1 mechanism is.
Vice versa, if the carbocation formed in step 1 is very unstable, it will be formed in very small amounts, making the rate of step 1 rather slow. This makes it difficult to proceed to step 2 of the Sₙ1 mechanism. In this case, the Sₙ2 mechanism will be favoured instead, as there is no need for a carbocation to be formed in Sₙ2.
So, in deciding if any molecule undergoes Sₙ1 or Sₙ2, we just have to evaluate the stability of the carbocation.
Back to the question – Will 1–(chloromethyl)benzene undergo Sₙ1 or Sₙ2?
Let’s examine the stability of carbocation X. The positively-charged carbon has 3 regions of electron density around it (don’t forget the C–H bonds!) and is thus sp² hybridised. Every carbon in benzene is also sp² hybridised, thus we can draw the p orbitals in:

Since
1. All the p orbitals are adjacent to each other and
2. All the p orbitals are aligned to each other,
The empty p orbital of the positively charged carbon can overlap with the six p orbitals of benzene (pi electron cloud), thus allowing pi electrons from benzene to delocalise into the empty p orbital of the positively charged carbon. This disperses the positive charge, making the carbocation more stable.

Some of you might ask – how do we know that the pi electrons delocalise from the benzene ring to the positively-charged carbon and not the other way, from the positively-charged carbon into the benzene ring?
It can only be the former, because the p orbital on the positively charged carbon is empty and does not contain any electrons.

In step 1, electrons move from the C–Cl bond to Cl, so carbon loses electrons. Since carbon loses electrons, its p orbital will now be empty.
Think of resonance as bringing electrons from a region with excess electrons into a region deprived of electrons. An example would be moving electrons from a negatively charged region to a positively charged region.
In this carbocation, the positive charge on the carbon simply indicates a lack of electrons, or a region deprived of electrons. Therefore, electrons should move from the electron-rich pi electron cloud to the positive charge, filling the “gap/hole” there.

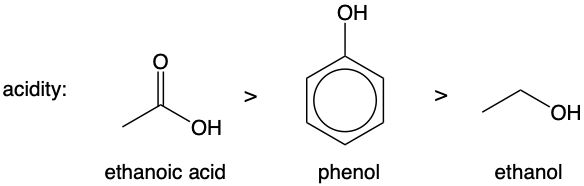
3. Acidity
What determines how acidic an acid is?
An acid HA dissociates in water according to the following equation:
HA ⇌ H⁺ + A⁻
The more stable A⁻ is, the more the position of equilibrium shifts to the right, producing more H⁺. Therefore, the more stable the conjugate base (A⁻), the more acidic its acid is.
Let us compare the acidity of these three molecules:

We should first draw out their conjugate bases:

Note the names of the conjugate bases (ethoxide, phenoxide, ethanoate) so that you can refer to them easily in your explanations.
The more stable the conjugate base, the more acidic its acid is, and hence we should compare the stabilities of these three conjugate bases.
The ethoxide is the least stable conjugate base due the electron- donating alkyl group which increases electron density on O, thus intensifying its negative charge.
In phenoxide, the O is sp² hybridised (it has at least one lone pair of electrons and is connected to a sp² hybridised carbon). Since O and all the carbons in benzene are sp² hybridised, they all have unhybridised p orbitals:

The p orbital on O can overlap with the pi electron cloud of the benzene ring, allowing the lone pair of electrons on O to delocalise into the benzene ring, dispersing the negative charge on O, making phenoxide more stable than ethoxide.
Try to visualise how the new pi electron cloud looks like. Since O is contributing its lone pair of electrons into the pi electron cloud, the original pi electron cloud of 6 delocalised electrons expands into a pi electron cloud of 8 delocalised electrons.

In ethanoate, the O⁻ is sp² hybridised (it has at least one lone pair of electrons and is connected to a sp² hybridised carbon), thus it has a p orbital. The C and O in the C=O bond must also have a p orbital each, as the p orbitals side-on overlap to form the C=O pi bond. Drawing in the p orbitals:

Remember that resonance is the movement of electrons from a region with excess
electrons to a region lacking electrons.

Since the ethanoate carbon is connected to two highly electronegative oxygen atoms,
it gains a partial positive charge. Electrons therefore flow from the negatively charged
O (excess electrons) to the electron-deficient carbon.
To describe this accurately, we say that the p orbital on O⁻ overlaps with the p orbitals
of C and O in C=O (the pi electron cloud of C=O), allowing the lone pair of electrons
on O⁻ to delocalise into the pi electron cloud of C=O. This disperses the negative
charge on O, stabilising the ethanoate ion.
Note that pi electron clouds do not necessarily have to be a benzene pi electron cloud.
C=O is also a pi electron cloud, a relatively small one that contains 2 pi electrons.
We seem to have stumbled upon a new conundrum here – Since delocalisation occurs for both the phenoxide and carboxylate ion, how do we know which is more stable?
We have to compare the extent of delocalisation in both ions (see diagram below). In ethanoate, the extent of delocalisation of the lone pair of electrons on O⁻ is greater as the electrons are attracted to the other highly electronegative O. Imagine the electrons being pulled up, towards the O of the C=O bond. In phenoxide, the extent of delocalisation of the lone pair of electrons on O⁻ is not as great as carbons are not as electronegative, hence pulling the electrons towards the benzene ring to a smaller extent.

Since delocalisation of the lone pair of electrons on O⁻ occur to a greater extent for ethanoate, the negative charge is more dispersed, making ethanoate more stable than phenoxide.
Ranking the stabilities of the conjugate bases, we have:

The more stable the conjugate base, the more stable. Therefore, ranking the acidities of the acids:
4. Basicity
What determines how basic a base is?
Let’s recall how a base works. There are two ways a base can work:
When NH₃ reacts with H⁺, it acts as both a Brønsted-Lowry base and a Lewis base as it accepts a proton and donates a lone pair of electrons to H⁺. In fact, for any base to react with a proton, it must donate its lone pair of electrons to the proton. For this reason, every Brønsted-Lowry base is a Lewis base. We can say that Brønsted-Lowry bases are a subset of Lewis bases.
However, not all Lewis bases are Brønsted-Lowry bases. When NH₃ reacts with BF₃, it donates its lone pair of electrons to B, thus acting as a Lewis base. However, it is obviously not a Brønsted-Lowry base as it is not accepting a proton here.

Either way, whether it acts as a Brønsted-Lowry base or a Lewis base, NH₃ has to donate its lone pair of electrons to act as a base. The more available the lone pair of electrons on N, the more likely it will be able to donate its lone pair, making it more basic.
Therefore,
the more available the lone pair of electrons to coordinate with an acid, the more basic the base.
Let us compare the basicity of these three molecules:

ethylamine: The electron-donating alkyl group increases the electron density on N, making the lone pair of electrons on N more available for coordination with an acid.
*Note: In phenylamine and ethanamide, the N is not fully sp² hybridised, but is somewhere between sp² and sp³ hybridised (out of syllabus). To be scientifically accurate, we will avoid saying that N is sp² hybridised/ has a p orbital.
phenylamine: The orbital* containing the lone pair of electrons on N overlaps with the pi electron cloud of benzene, allowing the lone pair of electrons to delocalise into the benzene ring. This decreases the availability of lone pair of electrons on N to coordinate with an acid, making it less basic than ethylamine.

ethanamide: The orbital* containing the lone pair of electrons on N overlaps with the pi electron cloud of C=O, allowing the lone pair of electrons to delocalise into C=O. This decreases the availability of lone pair of electrons on N to coordinate with an acid, making it less basic than ethylamine.

Is the extent of delocalisation greater in ethanamide or phenylamine? For that, we can employ the same logic as we did in point 4 of acidity.
In ethanamide, the extent of delocalisation of the lone pair of electrons on N is greater as the electrons are attracted to the highly electronegative O. Imagine the electrons being pulled up, towards the O of the C=O bond. In phenylamine, the extent of delocalisation of the lone pair of electrons on N is not as great as carbons are not as electronegative, hence pulling the electrons towards the benzene ring to a smaller extent.
Since the extent of delocalisation in ethanamide is greater than that in phenylamine, the lone pair of electrons on N is less available in ethanamide than phenylamine. In fact, the extent of delocalisation in an amide is so great that amides like ethanamide are essentially neutral.
Ranking their basicities, we have:

This concludes the four main applications of resonance in the H2 Chemistry syllabus! 😊
Drawing resonance structures
It is important to learn how to draw the resonance structures for some simple molecules, such as ethanoate. Let’s work through the thought process for ethanoate together.
Remember – electrons usually move from a region of excess electrons to a region deficient in electrons! Therefore, in diagram 1, we will move the lone pair of electrons from electron-rich negatively charged O towards the electron-deficient partial positively charged carbon.

In moving the lone pair of electrons from O to form a C–O pi bond, the ethanoate carbon will now have five bonds, which is not possible. Carbons should maximally have four bonds as carbon is in period 2 and cannot expand its octet. To prevent five bonds from forming, the pi bond of C=O breaks, as shown by the pink arrow in diagram 2. To determine where the pink arrow goes, remember that O is highly electronegative, pulling electrons from the pi bond of C=O towards itself. And there you have it! Diagram 2 is the correct way of drawing arrows and the resonance structure of ethanoate.
Students often make the following mistake, drawing the arrow to the carbon.

The arrow should point to the bond, not directly to the carbon atom! Remember that the arrow represents the movement of two electrons. The two electrons should move (blue arrow) to form a new C=O pi bond, whereby the two electrons (blue) are shared between C and O. It should not be the case where both blue electrons go to only C and C hogs both electrons.

Another way of thinking is this – in the C=O bond, the pink electron was contributed by C and the green electron was contributed by O. When the pink arrow moves both electrons to O, carbon loses only one electron (the pink electron). If we drew the blue arrow such that it lands directly on C, that would imply compensating C with two electrons! C only needs one electron. Therefore, draw the blue arrow to the bond, compensating C with only one blue electron.
コメント